Project Overview
Protein structure prediction is a critical issue in the field of life sciences, which is of great significance for understanding protein function and various life activities. Currently, semi-parametric deep-learning solutions such as AlphaFold2 have achieved comparable accuracy to experimental techniques such as cryo-electron microscopy. However, due to the heavy reliance on input homologous sequence information, the model has significant limitations in practical scenarios.

AIRFold, built on the foundation of AlphaFold2, aims to provide scalable, systematic solutions for the critical issue of protein structure prediction in the field of life sciences. AIRFold's unique Homology Miner module focuses on the mining and extraction of co-evolutionary information, intelligently and automatically extracting, analyzing, and processing the co-evolution information within protein homologous sequences (MSA). In addition, AIRFold offers a systematic structural prediction solution, integrating various leading structural prediction models such as AlphaFold2, RoseTTAFold2, single-sequence structure models like OmegaFold and ESMFold, and ultimately ranking and screening all predicted structures using a model quality estimation (MQE) module. To fully integrate these different modules, we provide a microservices architecture along with user-friendly APIs and a web-based graphical interface, making it convenient for developers and biochemical researchers to use our platform for structural prediction.。
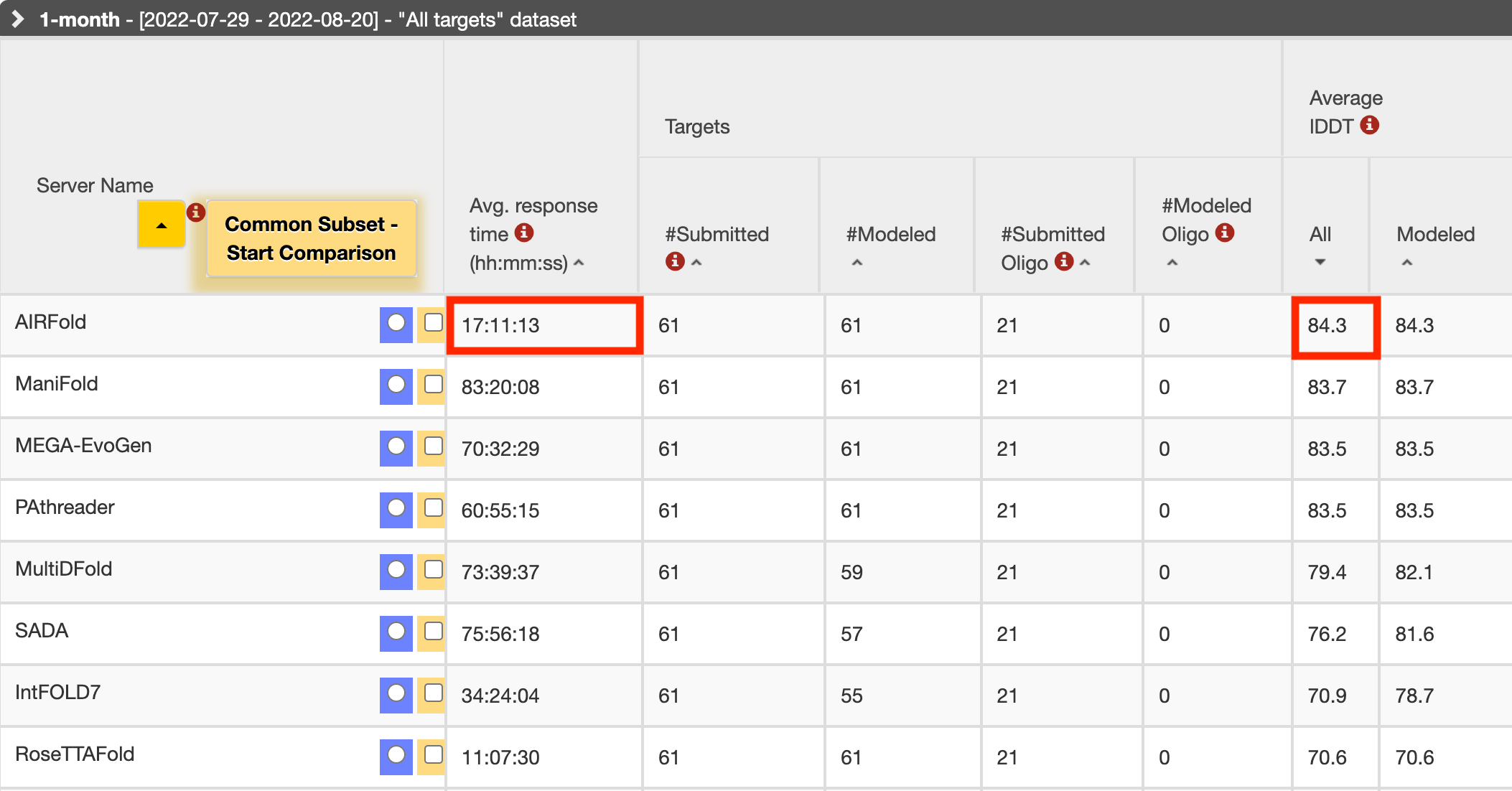
Research progress
AIRFold has won the global first place for four consecutive weeks in the authoritative protein structure prediction competition CAMEO. The team has built a fully automated control platform, including modules such as homologous sequence augmentation, homologous sequence selection, feature processing, structure prediction, result analysis, and automatic submission, which is much faster than other teams in terms of system response time. Specifically, in terms of the 'Hard' protein sequence, AIRFold is far ahead of second place.
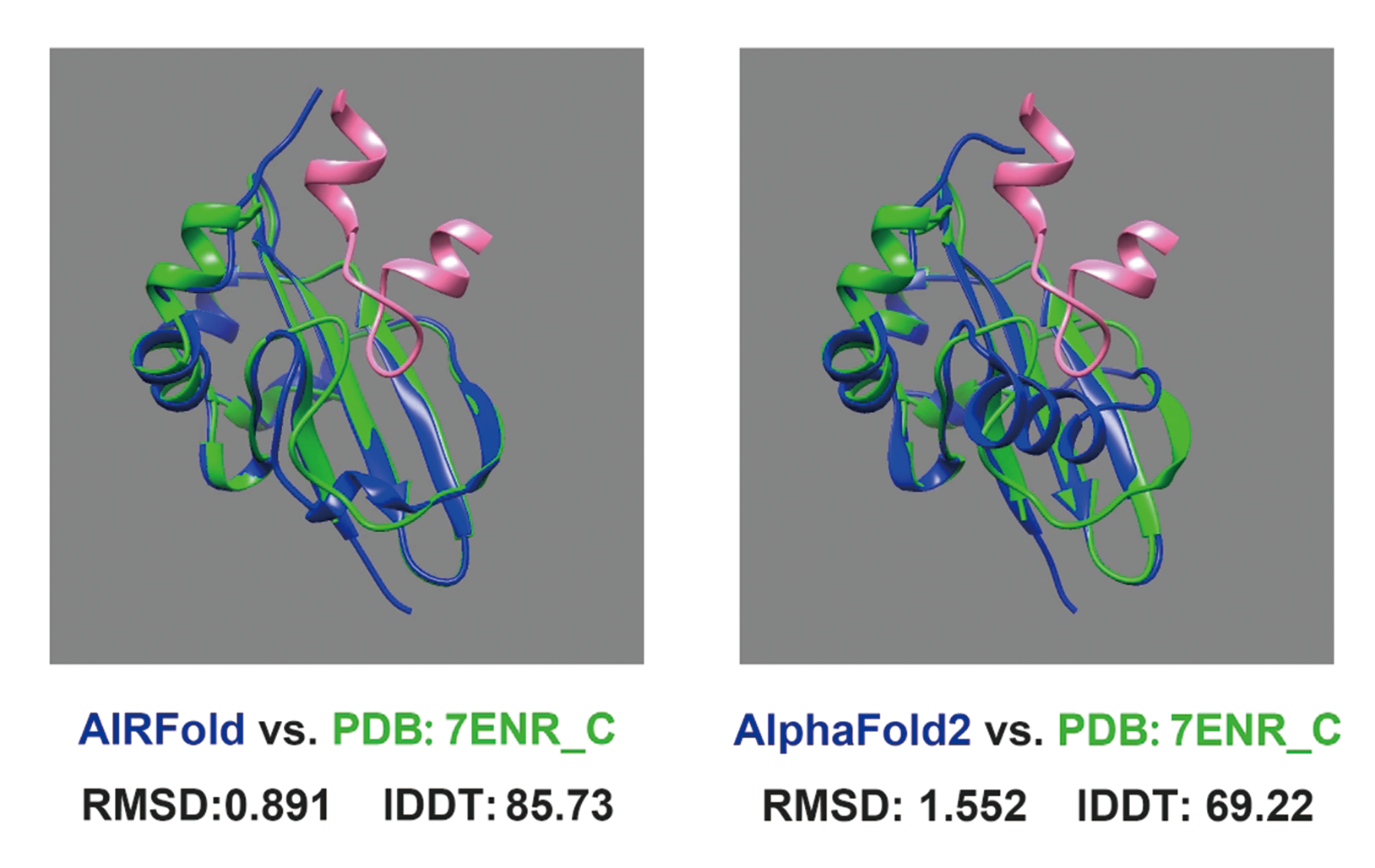
For example, for the protein with PDB number 7TVI, which comes from Cas13bt3, this protein has multiple domains, large conformations, and flexible conformations. The quality of homologous sequences obtained from multiple sequence alignment is relatively low. However, through the screening of homology mining modules, the signal-to-noise ratio of long-range interaction information in high-quality homologous sequences has been improved, and the relationship between multiple domains has been more accurately modeled. The results of AlphaFold2 are significantly better than those of Helical-1 and Helical-2, which mainly recognize crRNA (pink part) domains.

Future of project
The prediction of individual proteins and a deep understanding of coevolutionary information are the foundation for the team's future research on proteins and macromolecular drugs. AIRFold focuses on how protein structure determines its function and how it contributes to the development of drugs and therapies, rather than just the isolated issue of structural prediction. Therefore, the team is continuing to explore pharmaceutical related issues such as protein point mutations and multi conformation prediction, and is in close communication with relevant enterprises and research institutions. We look forward to more outstanding researchers joining this emerging interdisciplinary field in the future to further leverage the value of AI.